

What We Learned From The Trial of Our First AI-Discovered Drug
In the right neighborhood, but not yet the right house

In late 2025, Verge completed a Phase 1b clinical trial for VRG50635, a new investigational drug for patients living with ALS. The drug missed its primary efficacy endpoint, and we’re not advancing the program in ALS.
When a drug doesn’t work, the temptation is to look away. We’re going to do the opposite. The trial taught us more than a successful one might have, and we want to share what we learned.
Our first generation discovery engine, CONVERGE 1.0, nominated a drug target called PIKfyve. Our team took it through years of preclinical work, Phase 1 healthy-volunteer studies, and a Phase 1b study in ALS patients. The drug had clear biological activity, including in the brain and the two cell types most central to ALS, neurons and astrocytes. But the patients did not get better. Inside the gap between those two facts, we learned things about the disease and our platform that change how we build the next iteration.
We got very close. We were in the right neighborhood. We just have not yet found the right house yet. Here is what we learned, and what we are going to do with it.
What we set out to do
PIKfyve is an enzyme that helps cells take out the trash. It is part of the endolysosomal system, the machinery cells use to move, recycle, and dispose of internal cargo. From a multi-omic analysis of nearly 1,000 tissue samples from ALS patients and healthy controls, CONVERGE flagged PIKfyve as a top target. In ALS brain tissue, the gene network around PIKfyve was profoundly dysregulated. Motor neurons in ALS could no longer clear the protein aggregates piling up inside them, leading to cell death. The house was full of trash. Restoring the pathway should help take it out and keep the neurons alive longer.
In motor neurons grown from ALS patient stem cells, PIKfyve inhibition cleared protein aggregates and improved survival. In a mouse model of ALS, it reduced neurofilament, the same blood marker we’d later use to measure efficacy in the clinic. Independent labs saw similar effects across multiple preclinical models (Shi et al., Nature Medicine, 2015; Hu et al., Cell, 2023). The biology was novel, the genetics were supportive, and the case for translation was strong.
We developed VRG50635, a potent, brain-penetrant PIKfyve inhibitor, and ran Phase 1 healthy-volunteer studies. We concluded that the drug was safe and well-tolerated. We identified plasma GPNMB as a reliable pharmacodynamic biomarker: a readout confirming the drug was hitting the PIKfyve pathway. We then ran a Phase 1b in 54 patients with ALS powered to detect efficacy, which we defined as a 30% drop in a protein called plasma neurofilament light chain (NfL).
NfL is a protein neurons release as they die. A fall in NfL levels in the blood is widely read as neurons dying more slowly, which should translate into slower disease progression. It is a widely used biomarker of efficacy in ALS clinical trials.
Pharmacologically, the drug behaved as designed. It reached the brain at concentrations roughly twice what’s needed to fully inhibit PIKfyve. GPNMB in blood and cerebrospinal fluid (CSF) rose in proportion to drug exposure: precisely the response one would expect if the drug were hitting its target.
Then the data showed something we did not anticipate.
The two surprises
Plasma NfL went up, not down
We had assumed plasma NfL would fall once cellular trash clearance turned on. Instead, plasma NfL rose. The signal was detectable within 2 weeks of starting the drug and stayed elevated as long as patients were on it. Plasma GFAP, another protein from inside astrocytes, climbed six hours after the first dose and tracked with NfL throughout the trial.
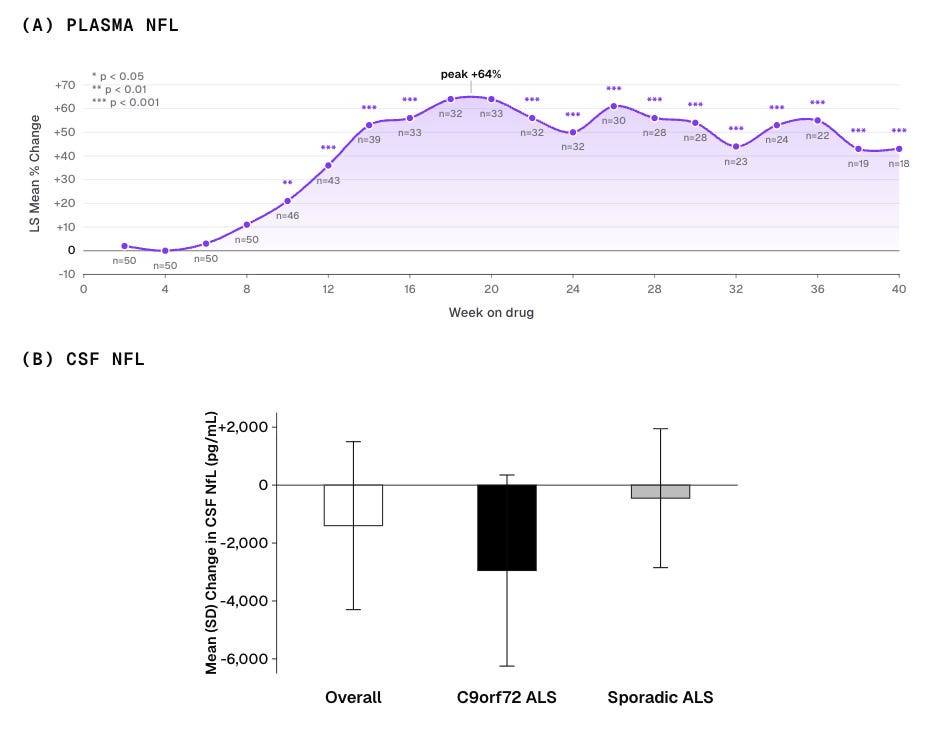
Spinal fluid told a different story. CSF NfL trended down, with a larger drop in patients with the C9orf72 genetic form of ALS. A reduction in NfL in CSF is considered early evidence of clinical benefit. But the primary efficacy endpoint was a reduction in plasma NfL, and plasma did the opposite of what we had bet on.
We believe the rise in plasma NfL was a pharmacodynamic response to PIKfyve inhibition — a direct effect of the drug’s mechanism, not worsening disease. Patients on drug did not get worse faster, and when dosing was interrupted, plasma NfL fell rapidly, on a timescale that matches drug clearance, not recovery from neuronal injury.
The biology makes sense in hindsight. PIKfyve inhibition activates a cellular pathway (secretory exocytosis) that actively pushes intracellular contents out of the cell. The drug was opening the door to expel the trash inside. Misfolded proteins from neurons from inside diseased cells came pouring into the bloodstream, including NfL from neurons and GFAP from astrocytes. Plasma NfL was tracking the drug’s action, not the disease. Unfortunately, it was also our efficacy endpoint.
NfL, one of the field’s most relied-upon efficacy biomarkers, behaves in a mechanism-dependent way when an intervention engages the cell’s recycling system — a finding we did not anticipate. Given that the endolysosomal pathway is of high-interest in many neurodegenerative diseases, it’s one we think the field should be aware of.
Without a reliable efficacy biomarker, we had to rely only on clinical endpoints, where no effect was seen within the constraints of the trial.
ALS patients behaved differently
Our first Phase 1 healthy volunteer study was uneventful. In healthy people, the drug was well tolerated at all doses, except at very high exposures.
Then came the Phase 1b trial in ALS patients. To our surprise, a third of ALS patients could not tolerate even the lowest dose and discontinued before any dose escalation. But the other two-thirds escalated through higher doses and looked much like the healthy volunteers. Why?
The split tracked with disease severity. Early discontinuers tended to be older, with more advanced ALS and worse prognostic scores. They responded differently to the same drug despite identical pharmacology. Something in their underlying biology was driving the split.
We measured 48 baseline lab values and biomarkers. Two — and only two — were significantly different between the two groups: plasma NfL and plasma GFAP. Both were elevated at baseline in the patients who did not tolerate the drug. The same two proteins our drug was pushing into circulation.
Severe ALS patients here seem to be doing the PIKfyve thing on their own. Their cellular recycling system is already running flat out, presumably as a response to the misfolded protein piling up in their neurons. We came in with a drug that pushed the same dial the disease was already pushing. For the sickest patients, there was no dial left to turn, only side effects.
A similar pattern shows up in immunology. IL-10 is one of the most potent anti-inflammatory cytokines in the body. It has never been approved as a therapy, partly because acutely sick patients are already producing high levels of IL-10 on their own. The pathway is saturated. The drug has no room to work.
PIKfyve dysregulation is not a yes-or-no feature of ALS; it tracks with how sick the patient is. Some patients are at the start of that arc, some are near the end, and they likely need very different drugs, very different doses, or possibly the opposite intervention entirely. We treated them as one population. The trial taught us they are not.
What the platform got right
Even though the drug did not deliver clinical benefit, it’s worth examining where we were able to move the needle.
The drug moved real biology in the cells the disease actually lives in. Most ALS trials end with no biological signal at all. In ours, the drug crossed the blood-brain barrier, engaged the pathway, and triggered rapid release of NfL and GFAP. Since NfL comes from neurons and GFAP from astrocytes, the drug was clearly triggering biological activity within the two cell types most central to ALS.
We could prove the drug reached its target, something many past ALS trials lacked. In our PIKfyve network from human tissue, GPNMB stood out as one of the most tightly coupled genes to PIKfyve. In ALS patient blood and spinal fluid, we found GPNMB was released by cells, detectable in both, and elevated in disease. The drug raised GPNMB across cells, ALS patient-derived motor neurons, and rodents, in lockstep with how much drug was on board. In healthy volunteers, the same pattern held in blood and spinal fluid (Gontier et al., Clinical and Translational Science, 2026). That gave us a clear readout that the drug was hitting its target in both the body and the brain before we dosed a single ALS patient, and a way to set the right dose for the Phase 1b.
The platform predicted new ALS-specific biology. In the clinic, the PIKfyve pathway turned out to be profoundly different not only between ALS patients and healthy volunteers, but within ALS itself. The sicker the patient, the more their cells had already activated the PIKfyve pathway in response to the disease, and the more strongly they reacted to the drug. This points to a new, disease-relevant biology: in ALS, cells appear to turn up the PIKfyve pathway as a compensatory defense against the disease, with the response scaling alongside severity.
Finding biology that tracks with a disease is one thing. Knowing which patients to treat, and when, is another. That is the gap CONVERGE 1.0 left open, and the one CONVERGE 2.0 is built to close.
What the platform missed
The trial taught us what our first iteration of the platform missed, and what is needed to build a transformational one.
The first is resolving individual patient heterogeneity. CONVERGE 1.0 was trained mostly on bulk post-mortem brain tissue, optimized to recover the average ALS-vs-control signal. It missed that the PIKfyve pathway sits in a fundamentally different state in a patient with mild disease versus advanced disease. We learned that the hard way: a third of ALS patients did not tolerate the drug.
The second is bridging dead tissue to living people. The only molecular data we could collect from patients during the trial was peripheral, mostly blood. CONVERGE was trained on brain tissue. We had no way to connect blood signatures back to brain biology, and so no way to characterize an individual patient’s underlying state from the samples we could actually collect.
The third is predicting how biomarkers and clinical endpoints will behave once a drug is given. CONVERGE did not predict that plasma NfL would rise in response to PIKfyve inhibition, and neither did our preclinical models. A platform that identifies a disease-relevant network is not the same as a platform that can simulate the readouts used to judge whether the drug worked.
Modern generative AI, built on the right data, can now address each of these gaps.
What’s next and why now?
ALS is one of the hardest diseases in drug development. Even if a platform doubled the industry’s clinical success rate (a transformational result), more than 80% of programs would still fail. We are not guaranteeing success on any single asset. We are raising the probability of success across a portfolio.
Transformational platforms are not built overnight. They come from persisting after setbacks and iterating on hard-won lessons. We have done what many companies in our space have not: taken a novel target from an AI platform through to a clinical proof of concept trial with clear biological activity. Along the way, we accumulated what we believe is the largest proprietary, curated neuro tissue dataset in the world — more than 12,000 human tissue samples across 6,000 patients and 15 million single-cell profiles, paired with clinical and genetic annotations across ALS, FTD, PSP, Parkinson’s, schizophrenia, and others. The PIKfyve trial now adds one of the most thoroughly phenotyped clinical and biomarker datasets ever generated in ALS.
What has changed in the past two years is the AI itself: recent breakthroughs in architecture have arrived just as our dataset has reached the scale they need. The hardest problem in neuroscience data is its incompleteness. Brain tissue is the molecular ground truth for neurodegenerative disease, but it can only come from autopsy patients. From living patients we get accessible proxies for the brain: blood, imaging, and clinical data. The two halves of the picture come from different patients.
Modern generative AI can bridge that gap. It learns relationships across modalities from many patients and infers what is missing. But the model only works if it has brain tissue to anchor those relationships. Without it, the proxies show only the disease’s downstream effects, with no way to trace them back to what is actually driving the disease in the brain. Our decade of brain tissue data is what makes the architecture work: it lets us link brain biology, longitudinal blood, and clinical trajectory into a single picture of each patient, and reason about how that patient would respond to a given drug. That is the class of questions the PIKfyve trial asked and we could not answer.
CONVERGE 1.0 was a discovery tool, built to predict novel targets from human data in the toughest diseases. We are now expanding CONVERGE 2.0 to include a precision neuro model, by training the latest generative AI architectures on a decade of multimodal neuroscience patient and clinical data. The output is what we call a virtual biopsy of the brain — an inferred molecular picture of a patient’s brain, generated from a routine blood draw. From there, it is designed to match patients to the right therapies, predict how patients will evolve over the course of their disease, and predict how biomarkers will respond to different interventions.
In other words, CONVERGE 2.0 is designed to catch exactly what CONVERGE 1.0 missed. Patient heterogeneity, identified from a baseline blood draw before any patient is dosed. Brain state, inferred from the peripheral biomarkers we can actually collect from living patients. And the direction a biomarker will move in response to a given intervention, predicted in advance rather than discovered after the trial reads out.
Together, these capabilities let us predict not only which targets to drug, but which patients to drug them in.
We were in the right neighborhood. Fortunately, the map is about to get considerably better.
This identification that the phase of disease matters is so important—thank you for sharing the learnings. I wonder if incorporating this principle into Alzheimer’s trials would have solved some of the failures there. What if GLP-1s were used in the compensatory window before microglial burnout? What if amyloid-targeted agents were used before it was measurable and the clearance pathway overwhelmed?
A very interesting piece; there is so much potential insight to be garnered regarding identifying the underlying causes of disease heterogeneity. Away from Neurodegeneration, in epilepsy and psychiatry (depression) roughly one third of patients do not respond to current treatments. Would be very interesting to see if the team could help unlock the biological underpinnings in these examples.